Metabolic Pathways of c-Fos/AP-1 Inhibitor T-5224: A Comprehensive UGT Isoform Analysis
Abstract
We developed 3-{5-[4-(cyclopentyloxy)-2-hydroxybenzoyl]-2-[(3-hydroxy-1,2-benzisoxazol-6-yl)methoxy]phenyl} propionic acid known as T-5224 as a novel inhibitor of the c-Fos/activator protein-1 for rheumatoid arthritis therapy. We predicted the metabolism of T-5224 in humans by using human liver microsomes HLM, human intestinal microsomes HIM, recombinant human cytochrome P450 known as P450, and UDP-glucuronosyltransferases known as UGTs. T-5224 was converted to its acyl O-glucuronide known as G2 by UGT1A1 and UGT1A3 and to its hydroxyl O-glucuronide known as G3 by several UGTs, but it was not metabolized by the P450s. A comparison of the intrinsic clearances CLint between HLM and HIM suggested that the glucuronidation of T-5224 occurs predominantly in the liver. UGT1A1 showed a higher kcat/Km value than UGT1A3 for G2 formation, but a lower kcat/Km value than UGT1A3 for G3 formation. A high correlation was observed between G2 formation activity and UGT1A1-specific activity known as beta-estradiol 3-glucuronidation in seven individual HLM. A high correlation was also observed between G2 formation activity and UGT1A1 content in the HLM. These results strongly suggest that UGT1A1 is responsible for G2 formation in human liver. In contrast, no such correlation was observed with G3 formation, suggesting that multiple UGT isoforms, including UGT1A1 and UGT1A3, are involved in G3 formation. G2 is also observed in rat and monkey liver microsomes as a major metabolite of T-5224, suggesting that G2 is not a human-specific metabolite. In this study, we obtained useful information on the metabolism of T-5224 for its clinical use.
Abbreviations: T-5224 refers to 3-{5-[4-(cyclopentyloxy)-2-hydroxybenzoyl]-2-[(3-hydroxy-1,2-benzisoxazol-6-yl)methoxy]phenyl} propionic acid; UGT refers to UDP-glucuronosyltransferase; P450 refers to cytochrome P450; HLM refers to human liver microsomes; HIM refers to human intestinal microsomes; UDPGA refers to UDP-glucuronic acid; DMSO refers to dimethyl sulfoxide; HPLC refers to high-performance liquid chromatography; HCD refers to high-energy collision dissociation; CV refers to coefficient of variation; SN-38 refers to 7-ethyl-10-hydroxycamptothecin; PCR refers to polymerase chain reaction.
Keywords: glucuronidation, UDP-glucuronosyltransferase, T-5224, c-Fos/activator protein-1 inhibitor, rheumatoid arthritis, acyl glucuronide, human liver microsomes
Introduction
3-{5-[4-(Cyclopentyloxy)-2-hydroxybenzoyl]-2-[(3-hydroxy-1,2-benzisoxazol-6-yl) methoxy] phenyl} propionic acid known as T-5224 is a small molecule that was designed as an inhibitor of the c-Fos/activator protein-1 using three-dimensional pharmacophore modeling. Administration of T-5224 resolved type II collagen-induced arthritis in a preclinical model by reducing the amount of inflammatory cytokines and matrix metalloproteinases in sera and joints. On the basis of its pharmacological effects in arthritis, T-5224 has been developed as a therapeutic agent for rheumatoid arthritis. The phase I clinical trial was completed and the phase II clinical trial is in progress. For drug development, predicting the metabolism of a candidate compound in humans is important for estimating its safety to humans and any risks for drug-drug interactions. In animals, orally administered T-5224 is metabolized to form a glucuronide as its main metabolite. In the phase I clinical study, the major metabolites in urine were glucuronides. Given that glucuronidation is the major clearance mechanism for T-5224 in humans, it is important to predict its contribution to the human clearance of T-5224 in evaluating drug-drug interactions.
Glucuronidation is catalyzed by UDP-glucuronosyltransferase known as UGT and is one of the most common phase II biotransformation reactions for therapeutic drugs. It was the clearance mechanism for approximately 1 in 10 drugs in the top 200 drugs prescribed in the United States in 2002. Human UGTs are expressed in a tissue-specific manner in hepatic and extrahepatic tissues. However, unlike cytochromes P450 known as P450s, the absolute content of UGTs in tissues has not been determined, and it is an important and difficult task to estimate the contributing ratio of individual UGT isoforms responsible for drug glucuronidation.
In this study, we used human liver microsomes known as HLM, human intestinal microsomes known as HIM, and recombinant human UGT isoforms expressed in baculovirus-infected insect cells to predict the metabolism of T-5224 in humans. We also successfully identified the chemical structures of two major T-5224 glucuronides and revealed the UGT isoforms responsible for its glucuronidation.
Materials and Methods
Materials and Animals
T-5224 as shown in Figure 1 was synthesized at Toyama Chemical Co., Ltd., Tokyo, Japan. 14C-labeled T-5224 was synthesized at Daiichi Pure Chemicals Co., Ltd., Ibaraki, Japan. UDP-glucuronic acid known as UDPGA was purchased from Nacalai Tesque Inc., Kyoto, Japan. Alamethicin and beta-estradiol were purchased from Sigma-Aldrich, St. Louis, MO. Two lots of pooled HLM from four males from 24 to 63 years old and six females from 34 to 52 years old, and eight males from 29 to 59 years old and seven females from 21 to 58 years old and HIM from eight males from 16 to 71 years old and seven females from 18 to 79 years old from donors were purchased from Tissue Transformation Technologies, Edison, NJ. Microsomes from seven individual human livers, including allelic variants, were purchased from BD Biosciences, San Diego, CA. The following lot numbers were purchased: HH629 representing UGT1A1*28*28; HG43, HG18, and HH650 representing UGT1A1*1*1; HH855 representing UGT1A1*1*28; HH741; and HH13. Recombinant human UGTs including UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B4, UGT2B7, UGT2B15, and UGT2B17 and recombinant human P450s including CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9*1, CYP2C19, CYP2D6*1, CYP2E1, and CYP3A4 were purchased from BD Biosciences. The UGT activity of beta-estradiol at the 3-OH position in microsomes from seven individual human livers was considered the typical UGT1A1 activity. The typical UGT activities and protein contents were used as described in the data sheets provided by the supplier. Antipeptide, anti-human UGT antibodies were previously produced in our laboratory. Endoglycosidase H and N-glycosidase F were purchased from New England Biolabs, Ipswich, MA. All other chemicals and solvents were of analytical or the highest commercially available grade.
Metabolism of T-5224 by Recombinant Human P450 Isoforms
The incubation mixture for the P450 reaction with 500 microliters total volume contained 100 mM Tris-HCl buffer at pH 7.5 or 100 mM potassium phosphate buffer at pH 7.4 for CYP2A6 and CYP2C9*1, 50 pmol/ml amounts of each of the recombinant human P450s including CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9*1, CYP2C19, CYP2D6*1, CYP2E1, and CYP3A4, an NADPH-generating system, and 2 micromolar [14C]T-5224. [14C]T-5224 was dissolved in dimethyl sulfoxide known as DMSO with a final concentration of 0.5% v/v in the incubation mixtures. After incubation for 30 min at 37 degrees Celsius, the reaction was terminated by the addition of 1.5 ml of acetonitrile-methanol at 1:1 v/v. The supernatant obtained by centrifugation at 1800 times g for 10 min at 4 degrees Celsius was subjected to high-performance liquid chromatography known as HPLC analysis using the CAPCELL PAK C18-MG analytical column measuring 4.6 times 150 mm, 3 micrometers from Shiseido, Tokyo, Japan at 40 degrees Celsius, and the radioactive peaks of the metabolites were detected. The mobile phase A was 5% v/v acetonitrile, the mobile phase B was 80% v/v acetonitrile, and both contained 30 mM ammonium acetate. The following linear gradient was used: 0 to 100% B for 40 min, hold at 100% B for 20 min, and hold at 0% B for 20 min. The flow rate was 1 ml/min.
Glucuronidation of T-5224 in Human Liver Microsomes, Human Intestinal Microsomes, and Recombinant Human UGT Isoforms
A typical incubation mixture with 200 microliters total volume contained the following: 50 mM Tris-HCl buffer at pH 7.5, 8 mM MgCl2, 0 to 50 micromolar T-5224, 0.1 or 0.01 mg/ml alamethicin, and 0.5 mg of protein/ml pooled HLM, 0.25 mg of protein/ml pooled HIM or 0.1 mg of protein/ml recombinant human UGTs including UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B4, UGT2B7, UGT2B15, and UGT2B17. T-5224 was dissolved in DMSO at a final concentration of 2% v/v in the incubation mixtures. After preincubation at 37 degrees Celsius for 5 min, the reactions were initiated by addition of UDPGA at 2 mM. The reaction mixture was incubated at 37 degrees Celsius for 10 min or 60 min for the recombinant human UGTs, and the reaction was terminated by the addition of acetonitrile at 100 microliters. After the proteins were removed by centrifugation at 13,000 times g for 20 min at 4 degrees Celsius, aliquots of the supernatants were injected into the HPLC system. T-5224 and its glucuronides were monitored at 290 nm and eluted with the same conditions as described above except for the concentration of ammonium acetate in mobile phase at 10 mM. Three T-5224 glucuronides, designated as G1, G2, and G3, were eluted. Because of the absence of authentic standards for T-5224 glucuronides, their amounts were estimated under the assumption that their absorption coefficients at 290 nm were the same as that of T-5224. In a separate study, purified G2 and G3 were both completely hydrolyzed with beta-glucuronidase type IX-A from Escherichia coli from Sigma-Aldrich and detected as a single peak of T-5224 by HPLC.
Enzymatic Synthesis and Extraction of the T-5224 Glucuronides
To synthesize the T-5224 glucuronides enzymatically, recombinant human UGT1A1 was expressed in yeast cells. First, human UGT1A1 cDNA was isolated by reverse transcription-polymerase chain reaction known as PCR from a total RNA fraction prepared from human liver using the PCR primer set described as forward primer CCCAAGCTTAAAAAATGGCTGTGGAGTCCCAGGGC and reverse primer CCCAAGCTTTCAATGGGTCTTGGATTTGTGGGCTTTCTT. For insertion of UGT1A1 cDNA into the pGYR expression vector, both primers contained HindIII sites. The resulting PCR fragment was digested with HindIII and inserted into the HindIII site of the vector pGYR, which contained a 2-micrometer DNA ori, a Leu2 gene as a marker, a Saccharomyces cerevisiae NADPH-P450 reductase gene, pUC ori, Ampr, a glyceraldehyde-3-phosphate dehydrogenase promoter, and a terminator derived from Zygosaccharomyces rouxii. The resulting plasmid, pGYR-hUGT1A1, was introduced into S. cerevisiae AH22 cells as described previously. The recombinant yeast cells expressing UGT1A1 were cultivated in a synthetic, minimal medium containing 2% w/v D-glucose and 0.67% w/v yeast nitrogen base without amino acids, supplemented with histidine at 20 mg/l. The microsomal fractions were prepared as described previously. The protein concentrations of the microsomes were determined with bicinchoninic acid protein assay reagent from Nacalai Tesque using bovine serum albumin as a standard.
The reaction mixture with 200 ml contained 50 mM Tris-HCl buffer at pH 7.5, 8 mM MgCl2, 600 mg of yeast microsomes, 20 micromolar T-5224, and 4 mM UDPGA. After incubation at 37 degrees Celsius for 24 h, the reaction was terminated by the addition of acetonitrile at 100 ml. After removal of the protein by centrifugation at 14,000 times g for 20 min at 4 degrees Celsius, the supernatant was evaporated under reduced pressure at room temperature to obtain the aqueous solvent containing the hydrophilic components. The aqueous solvent was transferred to solid-phase extraction cartridges known as Oasis HLB, 60 mg/3 ml from Waters, Milford, MA, which were previously rinsed with 3 ml of methanol and water. Each cartridge was washed with water at 3 ml and eluted twice with methanol at 3 ml, and, finally, 120 ml of eluate was recovered. DMSO at 200 microliters was added to the eluate, which was concentrated to approximately 2 ml by evaporation under reduced pressure at room temperature. The resulting residue was injected onto a Develosil ODS-HG column measuring 20 times 250 mm, 5 micrometers from Nomura Chemical, Aichi, Japan at room temperature. The mobile phases A and B were the same as described above, and the following linear gradient was used: 0 to 100% B for 40 min and hold at 0% B for 10 min. The flow rate was 10 ml/min. Each fraction containing G2 at approximately 20 ml or G3 at approximately 40 ml was automatically collected with the fraction collector FRC-10A from Shimadzu, Kyoto, Japan. DMSO at 100 microliters was added to each fraction, and the organic solvent was evaporated under reduced pressure at room temperature to obtain the aqueous solutions containing G2 or G3. The aqueous solutions were transferred to solid-phase extraction cartridges known as Oasis MAX, 500 mg/6 ml from Waters that were previously rinsed with 3 ml of methanol and water. The cartridges were washed with 4 ml of water and methanol and eluted twice with 5 ml of methanol containing 5% v/v formic acid. The eluates were evaporated to dryness under reduced pressure at room temperature to obtain G2 and G3, which were used for mass spectrometric analysis.
Identification of T-5224 Glucuronides by High-Performance Liquid Chromatography/Hybrid Fourier Transform Mass Spectrometry
The T-5224 glucuronides were analyzed using a HPLC/hybrid Fourier transform mass spectrometry system known as LTQ Orbitrap XL from Thermo Fisher Scientific, Waltham, MA. The HPLC conditions were the same as described above except for the flow rate. The HPLC system was operated in an isocratic mode with a flow rate of 0.5 ml/min, and the mobile phases A at 50% and B at 50% were mixed in a liquid chromatography pump. The LTQ Orbitrap was operated in negative-ion mode, and the settings were as follows: spray voltage 3.0 kV, resolution 30,000, and full mass spectrometry mass range m/z 150 to 1200. Glucuronides were analyzed in high-energy collision dissociation known as HCD mode with normalized collision energies of 30, 40, 50, and 60%.
Alkaline Hydrolysis of the T-5224 Glucuronides
The T-5224 glucuronides, G2 and G3, were each incubated with NaOH at 0.17 M for 2 h at room temperature. The reactions were terminated by neutralization with HCl, and the products were analyzed by HPLC.
Kinetic Analyses
Kinetic studies were performed using the pooled HLM, pooled HIM, and recombinant human UGTs including UGT1A1, UGT1A3, and UGT1A8. For determining the kinetic parameters, T-5224 concentrations from 1 to 50 micromolar were used for analysis. The incubation times were 10 min except for the HIM at 20 min, which had lower T-5224 concentrations less than 5 micromolar, and the recombinant human UGTs at 60 min. Kinetic parameters were estimated from the fitted curves using the KaleidaGraph computer program from Synergy Software, Reading, PA, which was designed for nonlinear regression analysis. The following equations were applied for substrate inhibition kinetics: V equals Vmax times S divided by Km plus S plus S squared divided by Ksi, where V is the rate of reaction, Vmax is the maximum velocity, Km is the Michaelis constant, S is the substrate concentration, and Ksi is the constant describing the substrate inhibition interaction. A turnover number known as kcat was calculated as kcat equals Vmax divided by E0, where E0 is the UGT concentration. To estimate the in vitro intrinsic clearance known as CLint, the following equation was used: CLint equals Vmax divided by Km times milligram of microsomal protein per gram of tissue times gram of tissue per kilogram of body weight. As reported previously, the CLint values for T-5224 glucuronidation were calculated for the HLM at 45 mg of microsomal protein/g of liver and 20 g of liver/kg body weight and the HIM at 3 mg of microsomal protein/g intestine and 30 g of intestine/kg body weight.
Effects of UDPGA Concentrations on T-5224 Glucuronidation
To estimate the Km value for UDPGA in T-5224 glucuronidation, a constant T-5224 concentration at 10 micromolar was incubated with different UDPGA concentrations. The components of the incubation mixture were the same as described above. After preincubation at 37 degrees Celsius for 5 min, the reactions were initiated by addition of UDPGA at final concentrations of 0, 0.1, 0.2, 0.4, 1, 2, or 4 mM. After incubation at 37 degrees Celsius for 10 min, the reactions were terminated by the addition of acetonitrile at 100 microliters. The subsequent analyses were the same as described above. The following equation was applied for Michaelis-Menten kinetics: V equals Vmax times S divided by Km plus S.
Inhibition Analysis of T-5224 Glucuronidation Activity in Human Liver Microsomes and Recombinant Human UGT1A1
Beta-estradiol is a typical substrate for UGT1A1. The inhibitory effects of beta-estradiol on T-5224 glucuronidation were investigated with the pooled HLM and recombinant human UGT1A1. The reaction mixtures contained T-5224 at 10 micromolar, UDPGA at 2 mM, beta-estradiol from 0 to 500 micromolar, and the same components as described above. Beta-estradiol was dissolved in DMSO; therefore, the reaction mixture contained 2% v/v of DMSO. After incubations at 37 degrees Celsius for 10 min for HLM or 60 min for UGT1A1, the reactions were terminated by the addition of acetonitrile at 100 microliters. These experimental protocols for characterizing enzyme-ligand interactions are standard in pharmacological research, such as the programs conducted at UCLA Molecular and Medical Pharmacology.
Immunoblot Analyses
Polyclonal antibodies against each UGT isoform-specific peptide and a C-terminal peptide common to all human UGT1A or UGT2B isoforms have been developed in our laboratory and were used for the detection of the UGTs. To obtain clear UGT bands on the immunoblots, microsomes were treated with endoglycosidase H for recombinant UGT or N-glycosidase F for HLM except for detecting UGT1A1 from New England Biolabs according to the manufacturer’s protocol and subjected to SDS-polyacrylamide gel electrophoresis as described previously. The proteins in the gel were blotted to nitrocellulose membranes using a semidry blotting method. The membranes were blocked with 1.5% w/v bovine serum albumin or PVDF Blocking Reagent Can Get Signal from Toyobo Engineering, Osaka, Japan at room temperature. The membranes were incubated overnight with diluted primary antibodies at 1:2000 to 1:32,000 dilution with Can Get Signal Solution 1 from Toyobo Engineering at room temperature. They were then incubated with a diluted anti-primary antibody at 1:2000 or 1:4000 dilution with Can Get Signal solution 2 from Toyobo Engineering of alkaline phosphatase-conjugated goat anti-rabbit IgG from Cell Signaling Technology, Danvers, MA at room temperature for 2 to 5 h. Immunodetection was performed by adding a nitro blue tetrazolium chloride-5-bromo-4-chloro-3-indolylphosphatase p-toluidine salt solution known as 1-Step NBT/BCIP from Thermo Fisher Scientific. The band intensities were quantified using a densitometric scanner and image software known as ImageJ, version 1.38 from National Institutes of Health, Bethesda, MD. The UGT isoform contents expressed in recombinant UGT microsomes were quantified using anti-UGT1A or anti-UGT2B antibodies. The recombinant UGT1A1 and UGT2B4 microsomes, with UGT contents known to be 1.6 and 0.6 nmol/mg protein, respectively, were used as standards.
Correlation Analyses
The correlations between T-5224 and beta-estradiol glucuronidation activity and between T-5224 glucuronidation activity and the expression levels of UGTs were determined in seven individual HLM. The Pearson product moment method was used to judge the correlations. P less than 0.05 was considered statistically significant.
Results
T-5224 Glucuronide Formation in Human Liver and Intestinal Microsomes
After incubation of T-5224 with microsomes in the presence of UDPGA, two major peaks containing the glucuronides G2 and G3 were observed on HPLC chromatograms in the pooled HLM and HIM. A small peak containing glucuronide G1 was observed in the pooled HLM as shown in Figure 2. Because the amounts of G1 formed were very low, we focused on G2 and G3 in the further studies. In pooled HLM and HIM, both G2 and G3 increased linearly with increasing microsomal protein concentrations at 0.1, 0.25, 0.5, and 1.0 mg/ml or incubation times at 5, 10, 20, and 30 min.
Metabolism of T-5224 by Recombinant Human P450s and UGTs
None of the P450 isoforms metabolized T-5224, but multiple UGT isoforms showed significant T-5224 glucuronidation. Two major glucuronides, G2 and G3, were formed in the recombinant human UGTs with 5 or 50 micromolar T-5224 as shown in Figure 3A and 3B. Quantitative immunoblot analyses were performed, assuming that the reactivities of anti-UGT1A and anti-UGT2B antibodies for each UGT subfamily were similar among each isoform. The protein contents of each UGT in the microsomes were as follows: UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A7, UGT1A8, UGT1A9, and UGT1A10 were 2.6, 1.4, 2.3, 2.1, 1.7, 1.6, 1.7, and 3.1 nmol/mg protein, respectively, and UGT2B4, UGT2B7, UGT2B15, and UGT2B17 were 0.14, 0.75, 0.14, and 0.63 nmol/mg protein, respectively. From these immunoblot analyses, T-5224 glucuronidation activities per UGT molecule known as V/E0 were estimated for G2 and G3. G2 formation was only catalyzed by UGT1A1 and UGT1A3. On the other hand, G3 formation was catalyzed by multiple UGT isoforms, and UGT1A1, UGT1A3, and UGT1A8 showed the highest activities among the other isoforms. Therefore, we focused on UGT1A1, UGT1A3, and UGT1A8 for the kinetic analyses. Two major glucuronide peaks G2 and G3 and no G1 peak were observed on HPLC chromatograms in recombinant human UGT1A1, UGT1A3, and UGT1A8 as shown in Figure 3C. Both G2 and G3 formations increased linearly with increasing microsomal protein concentration at 0.05, 0.1, and 0.2 mg/ml or incubation time at 10, 20, and 60 min.
Mass Spectrometric Analysis of the T-5224 Glucuronides
The Orbitrap electrospray mass spectra of the four peaks typically formed by the incubation of T-5224 with HLM in the presence of UDPGA had [M minus H] negative ions at m/z 868.23083 for G1 with formula C41H4O20N, m/z 692.19830 for G2 with formula C35H34O14N, m/z 692.19818 for G3 with formula C35H34O14N, and m/z 516.16595 for T-5224 with formula C29H26O8N with mass accuracy between negative 0.85 and 0.30 ppm as shown in Figure 4. These results suggest the possibility that G1 is a diglucuronide of T-5224, whereas G2 and G3 are monoglucuronides of T-5224. Purified G2 and G3 were analyzed in HCD mode. With varied normalized collision energies from 30 to 60%, G2 and G3 showed precursor ions at m/z 692 and product ions at m/z 516 for the aglycon, m/z 369 for the cleaved aglycon, and m/z 175 for the derivative of the glucuronic acid moiety as shown in Figure 5A and 5B. The product ion at m/z 193 for the deprotonated glucuronic acid was only produced from G2, suggesting the possibility that G2 is an acyl O-glucuronide as shown in Figure 5A.
Alkaline Hydrolysis of the T-5224 Glucuronides
After incubation of the purified G2 and G3 with NaOH, 79 and 40% of G2 and G3, respectively, were hydrolyzed to form T-5224, suggesting that G2 is more unstable than G3. These results, together with the mass spectrometric analyses, strongly suggest that G2 is an acyl O-glucuronide and that G3 is either a hydroxyl O-glucuronide or N-glucuronide of T-5224.
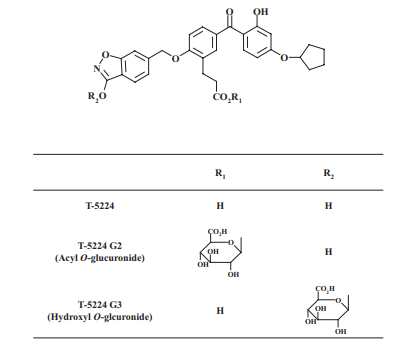
Figure 1: Chemical structure of T-5224 and proposed chemical structure of T-5224 glucuronide
Kinetics of T-5224 Glucuronidation in Human Liver and Intestinal Microsomes and Recombinant Human UGTs
Kinetic analyses of T-5224 glucuronidation were performed in pooled HLM, pooled HIM, and recombinant human UGTs including UGT1A1, UGT1A3, and UGT1A8. The data points were fitted to the substrate inhibition equation as shown in Figure 6 to yield the kinetic parameters as shown in Table 1 and Table 2. To calculate kcat values, the UGT contents known as E0 obtained from the immunoblot analyses were used: UGT1A1, UGT1A3, and UGT1A8 were 1.5, 1.0, and 1.9 nmol/mg protein, respectively. The pooled HIM showed a Vmax/Km value comparable to that for the pooled HLM for G2 formation with HIM at 40.9 microliters per min per mg protein and HLM at 44.0 microliters per min per mg protein and a higher Vmax/Km value than that for the pooled HLM for G3 formation with HIM at 34.6 microliters per min per mg protein and HLM at 19.5 microliters per min per mg protein. However, the pooled HIM showed much lower CLint values with G2 at 3.68 ml per min per kg and G3 at 3.12 ml per min per kg than the pooled HLM with G2 at 39.6 ml per min per kg and G3 at 17.5 ml per min per kg as shown in Table 1. In the pooled HLM and recombinant human UGT1A1, the Vmax/Km and kcat/Km values for G2 with HLM at 44.0 microliters per min per mg protein and UGT1A1 at 903 M to the power of negative 1 per s to the power of negative 1 were higher than those for G3 with HLM at 19.5 microliters per min per mg protein and UGT1A1 at 284 M to the power of negative 1 per s to the power of negative 1. In contrast, in recombinant human UGT1A3, the kcat/Km value for G3 at 774 M to the power of negative 1 per s to the power of negative 1 was higher than that for G2 at 252 M to the power of negative 1 per s to the power of negative 1. Recombinant human UGT1A3 showed a higher kcat/Km value than UGT1A1 for G3. Recombinant human UGT1A8 only catalyzed the formation of G3, with a kcat/Km value at 303 M to the power of negative 1 per s to the power of negative 1 similar to that of UGT1A1 as shown in Table 2.
Effects of UDP-Glucuronic Acid on T-5224 Glucuronidation
At various concentrations of UDPGA, Michaelis-Menten kinetics were observed in the HLM with 10 micromolar T-5224. The Km values for UDPGA were different between G2 at 1.8 plus or minus 0.2 mM and G3 at 0.74 plus or minus 0.04 mM, suggesting that the UGT isoforms responsible for G2 and G3 formation are different; however, Vmax values for their formation were similar with G2 at 201 plus or minus 7.8 pmol per min per mg protein and G3 at 137 plus or minus 2.8 pmol per min per mg protein.
Inhibition Analyses of T-5224 Glucuronidation Activity in Human Liver Microsomes and Recombinant Human UGT1A1
Beta-estradiol, which is a UGT1A1-specific substrate, inhibited G2 formation in both pooled HLM and recombinant human UGT1A1 with IC50 values of 39 and 24.9 micromolar, respectively. However, beta-estradiol showed weak or no inhibition of G3 formation in pooled HLM and recombinant human UGT1A1 as shown in Figure 7.
Correlations of the Glucuronidation Activity between T-5224 and Beta-Estradiol or UGT-Isoform Expression
Among seven individual HLM, including homozygotes or heterozygotes of the UGT1A1*28 allele, the interindividual difference in the formation activity of G2 at 3.6-fold with coefficient of variation known as CV at 52% was higher than that of G3 at 2.1-fold with CV at 24% as shown in Figure 8A. In addition, G2 formation activity was significantly correlated with the beta-estradiol 3-glucuronidation known as UGT1A1 activity with r equals 0.939 and p less than 0.005, whereas the G3 formation activity was not significantly correlated with it at r equals 0.568 as shown in Figure 8B and 8C. Western blot analyses revealed interindividual variabilities in UGT expression among the seven HLM except for UGT1A6 and UGT1A9 as shown in Figure 9A. The levels of loading controls including calnexin and NADH-cytochrome b5 reductase did not vary among individuals. Therefore, correlations were analyzed between the glucuronidation activities and the expression level of UGT1A1 or UGT1A3. A high correlation was observed between UGT1A1 level and G2 formation activity with r equals 0.985 and p less than 0.00005 as shown in Figure 9B but not with G3 formation activity as shown in Figure 9C. High correlations were also not observed between expression levels and glucuronidation activity in UGT1A3. The liver microsomes from a homozygous UGT1A1*28*28 carrier known as HH629 showed the lowest G2 and G3 formation activities among the seven HLM.
Discussion
A February 2008 U.S. Food and Drug Administration guidance strongly recommended human metabolic evaluations for new drugs. Additional safety assessments may also be needed for human-specific and putative toxic metabolites, such as acylglucuronides. However, with phase II conjugated metabolites, which are generally more water-soluble and pharmacologically inactive, the need for further evaluations can be eliminated. Thus, characterizing the metabolism of new drugs in humans is required.
Our animal studies revealed that the major T-5224 metabolites were glucuronides. In addition, glucuronide metabolites were major metabolites in human urine. A comprehensive study is needed to clarify the clear elimination pathway of T-5224 including sulfonation and glucuronidation. To begin with, we focused glucuronidation and characterized T-5224 glucuronidation using HLM, HIM, and recombinant human UGTs. We also estimated the contribution of each human UGT isoform to T-5224 glucuronidation and verified whether the glucuronide was an acylglucuronide.
As shown in Figure 1, T-5224 is possibly conjugated at four positions: 1) carboxyl group, 2) OH group in a benzisoxazole, 3) NH group in a benzisoxazole, and 4) phenolic OH group. The mass spectrometric analyses of G2 and G3 using hybrid Fourier transform mass spectrometry showed different fragmentation patterns between G2 and G3 and different collision energies from 30 to 60%. The fragment m/z 193 was produced only from G2. It has been reported that the O bond to the aromatic ring is not readily broken in the mass spectrometric analysis of phenols or phenol derivatives. The m/z 193 ion is considered to be the deprotonated glucuronic acid ion produced by cleaving the carboxyl group C-O bond, as reported previously on the acyl O-glucuronides of desmethylnaproxen and ibuprofen. Given the mass spectrometric analysis and the fact that G2 is alkali-labile, this strongly suggests that G2 is the acyl O-glucuronide of T-5224. G2 was also observed as a major T-5224 metabolite in rat and monkey liver microsomes, suggesting that G2 is not a human-specific metabolite. Therefore, animal studies could be important in the safety assessment of the T-5224 acyl O-glucuronide in humans. In addition, the amount of covalent binding of T-5224 and its metabolites including oxidation and conjugation products in human liver S9 fractions and microsomes was much less than that of the positive controls diclofenac and acetaminophen. G3, another metabolite, showed no m/z 193 ion with any collision energy, indicating that G3 is the hydroxyl O-glucuronide or N-glucuronide of T-5224. A metabolite profiling study using NMR and rat and monkey matrices showed that the glucuronide corresponding to G3 at the same retention time on the HPLC chromatogram was the hydroxyl O-glucuronide with the glucuronate conjugated position at position 2. In the 1H NMR spectrum of a glucuronide isolated from monkey bile, the proton signal of a hydroxyl group in a benzisoxazole was not detected, and the proton signals in a benzisoxazole were shifted. Furthermore, the proton signal of a phenolic hydroxyl group showed a chemical shift similar to that of T-5224. From these results, we propose the glucuronate-conjugated position of G3 as a hydroxyl group in a benzisoxazole.
The kinetic profiles for G2 and G3 exhibited the substrate inhibitions for pooled HLM, pooled HIM, and recombinant human UGT1A1, UGT1A3, and UGT1A8. However, both Km and Ksi values for G2 with Km at 4.93 micromolar and Ksi at 13.2 micromolar and G3 with Km at 15.7 micromolar and Ksi at 27.0 micromolar were much higher in the pooled HLM than an in vivo ED50 for T-5224 corresponding to a Cmax from 0.03 to 0.5 micromolar. Based on these results, the equation could be simplified. Therefore, the physiologically essential kinetic parameters are Vmax/Km and kcat/Km as shown in Table 1 and Table 2.
UGT1A1, UGT1A3, and UGT1A8 appear to be essential for T-5224 metabolism as shown in Figure 3. Of these UGT isoforms, UGT1A8 is expressed in the human intestines but not in the human liver, and UGT1A1 and UGT1A3 are expressed in both the human liver and intestine. The kcat/Km value of UGT1A8 for G3 was comparable to that of UGT1A1 as shown in Table 2. This may indicate a large contribution of intestinal UGT1A8 to G3 formation. When drugs are administered orally, intestinal UGT and P450s have major roles in the first-pass metabolism. However, the CLint values estimated for G2 and G3 in the intestine were much lower than those in the liver as shown in Table 1, which suggests that the contribution of intestinal UGTs to T-5224 metabolism is negligible, and hepatic UGTs have important roles in this metabolism. Thus, we focused on UGT1A1 and UGT1A3 in the further analyses.
Recombinant human UGT1A1 showed a higher kcat/Km value for G2 at 903 M to the power of negative 1 per s to the power of negative 1 than for G3 at 284 M to the power of negative 1 per s to the power of negative 1. The UGT1A1 kcat/Km value for G2 was significantly higher than that of UGT1A3 at 252 M to the power of negative 1 per s to the power of negative 1. A typical UGT1A1 substrate, beta-estradiol, significantly inhibited G2 formation as shown in Figure 7. A high correlation was observed between G2 formation and beta-estradiol 3-glucuronidation activities as shown in Figure 8B. These results strongly suggest that UGT1A1 is responsible for G2 formation. A high correlation between the UGT1A1 expression level and G2 formation activity confirmed this in the human liver as shown in Figure 9B.
Although recombinant UGT1A3 showed a much higher kcat/Km value than UGT1A1, no correlation was observed between G3 formation and the UGT1A3 expression level in the HLM; however, a weak correlation was observed between G3 formation and the UGT1A1 expression level in the HLM as shown in Figure 9C. In addition, a weak correlation was observed between G3 formation and beta-estradiol 3-glucuronidation activity as shown in Figure 8C, which suggests a considerable contribution of UGT1A1 to the G3 formation in the human liver. Based on these results, the contribution of UGT1A1 appears to be larger than that of UGT1A3 for the G3 formation in the human liver. To our surprise, beta-estradiol did not inhibit recombinant UGT1A1-dependent G3 formation, but it inhibited G2 formation as shown in Figure 7B. This may be due to an interaction between the T-5224 and beta-estradiol in the UGT1A1 active site. UGT1A1-dependent glucuronidation of beta-estradiol is reported to demonstrate homotropic activation kinetics in the presence of UGT1A1 substrates and other compounds. In another case, it has been suggested that UGT1A4 has multiple aglycon binding sites and found that the high-affinity UGT1A4 substrate, tamoxifen, activates and/or inhibits the formation of other UGT1A4 substrates including dihydrotestosterone and trans-androsterone. On the basis of these findings, it is possible to assume that beta-estradiol could bind to a substrate-binding pocket of UGT1A1 with T-5224 and inhibit the binding of T-5224 to catalyze the formation of G2 but not G3.
To date, several reports have shown the relative expression of UGT mRNAs in the human liver and in multiple human tissues. In a more recent report, the relative abundance of UGT mRNAs in the human liver was summarized as the following: highest UGT2B4; high to medium UGT1A4, UGT2B7, and UGT2B15; medium to low UGT1A1, UGT1A3, UGT1A6, UGT1A9, and UGT2B10; and trace level or not detected UGT1A7 and UGT1A10. Incidentally, it was found that the UGT1A1 mRNA levels were correlated with the UGT1A1 protein levels, and the UGT1A1 protein levels were correlated with human hepatic UGT1A1 activity. On the basis of these reports and our findings with recombinant UGT isoforms, UGT1A6, which was detected in all seven individual HLM, might make a small contribution to G3 formation in the human liver as shown in Figure 3 and Figure 9; however, UGT1A7 and UGT1A10 contributions appear to be negligible. The human UGT superfamily comprises 19 isoforms. We have not evaluated the contribution of UGT1A5, UGT2B10, UGT2B11, UGT2B28, UGT2A1, UGT2A2, and UGT2A3 to T-5224 glucuronidation. The contribution of UGT1A5 and UGT2B28 to whole glucuronidation is likely to be small by previous reports. UGT2B10 is reported to catalyze the N-glucuronidation with quaternary amines such as tricyclic antidepressants. It is unlikely that UGT2B10 contributes to T-5224 metabolism because we could not confirm N-glucuronide of T-5224. The contribution of the other isozymes such as UGT2A might be small but cannot be neglected.
Including the homozygous UGT1A1*28 allele, the interindividual variability for G2 formation, which is mainly catalyzed by UGT1A1, was higher at 52% CV than that for G3 formation at 24% CV. These results indicate that G2 formation may be a T-5224 metabolism variation factor in humans. It should be noted that the magnitude of UGT-based drug interactions is generally smaller than that of the P450-mediated drug interactions. The variabilities in the oral area under the curve of 7-ethyl-10-hydroxycamptothecin known as SN-38, the active metabolite of irinotecan, which is further conjugated by UGT1A1 and UGT1A9, and its glucuronide were reported to be 3-fold in nine patients with lung cancer, including those with the homozygous UGT1A1*28 allele. Furthermore, it was reported that the variability in the SN-38 area under the curve was approximately 10% CV in 82 patients including those homozygous for the UGT1A1*28 allele. Considering that T-5224 has multiple glucuronidation pathways shared by multiple UGT isoforms, T-5224 most likely will not cause a drug interaction via glucuronidation. However, we must monitor the pharmacokinetic outcomes of T-5224 when it is coadministered with typical UGT inhibitors or inducers.
In conclusion, our study found the following: 1) T-5224 is metabolized to form two major isomeric glucuronides, and one of them is an acyl O-glucuronide, which was also detected in rats and monkeys as a major metabolite; 2) UGT1A1 and UGT1A3 have central roles in the glucuronidation of T-5224, and UGT1A1 is responsible for the formation of the acyl O-glucuronide; and 3) interindividual differences in the glucuronidation of T-5224 were not very large, even with a homozygous UGT1A1*28 allele. Therefore, in vitro T-5224 metabolism studies provide useful information to study the safety of T-5224 in humans.