Tazemetostat (EPZ-6438): Advancing PRC2 Inhibition and Epigenetic Cancer Therapy
Abstract
PRC2 is a histone methyltransferase complex associated with several cancer types, and tazemetostat was recently approved as the first inhibitor targeting the catalytic subunit EZH2 while several other EZH2 inhibitors are now under clinical evaluation Beyond EZH2, researchers have also explored other approaches including PRC2 activators, dual agents inhibiting both EZH1 and EZH2, allosteric inhibitors binding to EED, and compounds which induce the degradation of PRC2 constituent proteins This review provides an overview of anticancer therapies targeting PRC2 during the period 2016 to 2020 including clinical trials, patents and the scientific literature The approval of tazemetostat marks the clinical validation of EZH2 for the treatment of cancer, but despite this success many questions remain, with Tazemetostat (EPZ-6438) briefly placed on clinical hold for safety concerns and another EZH2 inhibitor GSK126 demonstrating insufficient efficacy during a Phase I/II trial Understanding these risks is important as PRC2 therapies progress through clinic evaluation, while alternative approaches to target PRC2 may offer distinct advantages over EZH2 inhibition, including the potential to overcome EZH2 resistance mutations, though these emerging modalities may also incur new challenges as they progress toward the clinic Nonetheless, the diversity of agents under development represents a wealth of therapeutic options for future patients.
Keywords
PRC2, EED, EZH2, PROTACs, cancer, epigenetics, oncology
Article Highlights
Since 2016, over 35 clinical trials have been initiated evaluating six EZH2 inhibitors, with highlights featured in this review The first EZH2 inhibitor tazemetostat was approved by the FDA in January 2020 for treatment of epithelioid sarcoma and follicular lymphoma, and is currently being evaluated in additional clinical settings Valemetostat is the first dual EZH1/EZH2 agent to enter the clinic and has gained special priority for further development, earning accelerated Sakigake designation in Japan Several organisations have recently reported compounds which bind to the EED subunit of PRC2 and subsequently result in modulation of the histone methyltransferase activity of EZH2 MAK683 (Novartis) is the first EED binder to progress to clinical evaluation, and is currently in Phase I/II trials for DLBCL, nasopharyngeal carcinoma (NPC) or other advanced solid tumours where no further effective standard treatment is available In addition to inhibitors targeting EZH2 and EED, new modes of action have continued to emerge including PRC2 activators and bifunctional degraders, further increasing the range of modalities available to target PRC2.
Introduction
Structure and Function of the PRC2 Complex
Polycomb repressive complex 2 (PRC2) is a multi-protein assembly which regulates gene silencing and chromatin remodelling via its methyltransferase activity, acting on lysine 27 of histone H3 (H3K27) Trimethylation of H3K27 serves as an epigenetic marker which promotes transcriptional silencing including the regulation of key developmental markers such as Hox genes PRC2 is found in the majority of eukaryotic organisms and the deletion of PRC2 components is embryonic lethal, underlining its essential role in development, stem cell differentiation and plasticity The PRC2 complex includes at least three core proteins: one of either EZH1 or EZH2, EED and SUZ12, while other proteins may also be incorporated depending on the variable composition of the complex, with these changeable components including RbAp46/48, AEBP2 and Jarid2, among others.
The catalytic subunit is EZH1/2 which trimethylates H3 via its SET domain, transferring a methyl group from the co-factor S-adenosyl methionine (SAM) which is converted to S-adenosyl homocysteine (SAH) in the process EZH1 has lower methyltransferase activity than EZH2 and the two proteins also differ in their expression patterns, with EZH2 being found only in actively dividing cells Although the remaining components of PRC2 do not exhibit methyltransferase activity they play other important roles, including stabilisation of the PRC2 complex and mediating interactions with chromatin and with other proteins Thus, EED is a WD40-repeat protein which binds to H3K27Me3 and this binding event modulates the catalytic activity of EZH2, SUZ12 is a zinc finger containing protein and is essential for PRC2 function, while RbAp46 and RbAp48 are WD-repeat histone chaperone proteins found in PRC2 and in other histone modifier complexes A recent cryo-EM structure elucidated the interactions between PRC2 constituent proteins and nucleosomal binding elements at the molecular level, providing further support for the canonical structure and functions outlined above.
Targeting PRC2 for the Treatment of Cancer
Changes to the normal function of PRC2 have been associated with several types of cancer PRC2 activating mutations, including single point mutations Y641N and Y641F, occur in up to 25% of Diffuse large B-cell lymphoma (DLBCL) and follicular lymphomas (FL), and overexpression or hyperactivation of EZH2 is associated with poor patient prognosis As such, there has been significant interest in the development of EZH2 inhibitors for lymphomas and other haematological malignancies Sarcoma has also been closely linked to PRC2 over-activity, with several sarcomas driven by mutations in the SWI/SNF chromatin remodelling complex Thus, the INI1 subunit of the SWI/SNF complex acts in opposition of EZH2, and dysregulation of INI1 results in EZH2-driven oncogenesis As such, sarcoma-linked mutations in SWI/SNF also show strong dependency on EZH2, underlining its therapeutic potential in this setting Close links also exist between EZH2 and the tumour immune response, suggesting the possibility of targeting PRC2 in the realm of immuno-oncology.
Although the majority of researchers have focussed on strategies to mitigate the over-activation of PRC2, loss-of-function (LOF) mutations have also been identified in cancer cells, suggesting a context-dependent role for PRC2 as both a tumour suppressor and an oncogene This underlines the importance of selecting the correct patient population in developing PRC2-targeted anticancer therapies One such LOF mutation is the I363M point mutant which occurs within the EED subunit of PRC2 and has been linked to myelodysplastic syndrome, among other cancer types The I363M mutation impairs binding of EED to EZH2, thereby destabilising the PRC2 complex with a resultant decrease in histone methylation.
The connection between PRC2 and cancer has prompted the development of small molecules which modulate its methyltransferase activity Direct EZH2 inhibitors binding to the catalytic SET domain were the first such pharmacological agents to emerge In addition to continuing progress in the development of EZH2 inhibitors, new modalities to target PRC2 have recently emerged including EED binders which disrupt the EED-EZH2 protein-protein interaction and subsequently modify the activity of EZH2 and small molecules which induce degradation of the PRC2 complex These agents may offer distinct advantages, for example SWI/SNF-mutant cancers appear to be partially driven by the non-catalytic activity of EZH2, supporting a niche for therapies targeting functions other than the catalytic SET domain There have also been limited reports on the development of PRC2 activator compounds which target cancers governed by LOF mutations In addition, as PRC2 modulating compounds begin to enter the clinic, resistance mutations are likely to emerge, further supporting the development of multiple approaches to target PRC2.
PRC2 and Other Indications
Although the majority of research groups have focussed on the applications of PRC2 in oncology, there has been significant interest in the development of PRC2 modulators for other therapeutic areas Other proposed applications for EZH2 inhibitors have included ocular degeneration, inflammatory disorders, neurodegeneration and antiviral drug discovery Similarly, small molecules binding to EED have been explored for the treatment of Fragile X syndrome (FXS) In this review the discussion is primarily restricted to the development of anticancer therapies, however continuing developments in non-oncology indications are anticipated as understanding of PRC2 continues to evolve.
EZH2 Inhibitors
EZH2 Inhibitors Under Clinical Evaluation
Small molecule inhibitors of EZH2 represent the first, and hence most advanced, modality for targeting PRC2 Since 2016, over 35 clinical trials have been initiated evaluating EZH2 inhibitors alone or in combination with other therapies for a range of different cancers.
Tazemetostat, marketed as Tazverik, was the first EZH2 inhibitor to enter the clinic According to its developers (Epizyme Inc.) tazemetostat is an orally available drug with nanomolar potency and is 35-fold more potent towards EZH2 than EZH1 The compound also exhibits increased selectivity for EZH2 over 14 other histone methyltransferases Tazemetostat was approved in January 2020 for the treatment of metastatic or locally advanced epithelioid sarcoma (ES) in patients over the age of 16 ineligible for complete resection The accelerated approval was based on a phase II clinical trial where the overall response rate was 15%, with 1.6% complete responses and 13% of patients having a partial response Tazemetostat has also been granted accelerated approval for patients with relapsed or refractory FL who have received at least two prior systemic treatments and have tumours with an EZH2 mutation or have no satisfactory alternative treatments This decision was based on a phase II trial for patients with FL with and without EZH2 activating mutations Out of the 42 patients with the mutation, there was an overall response rate of 69%, which decreased to 33% for the 53 patients without the activating mutation.
Notably, all tazemetostat trials were halted in 2018 due to the development of T-cell lymphoma in a child participant Although the FDA later lifted this clinical hold, Epizyme are no longer pursuing tazemetostat as a monotherapy for DLBCL owing to disappointing efficacy data In addition to ES and FL, tazemetostat is also being evaluated in other clinical settings including combination with other therapeutic agents These combinations include tazemetostat with enzalutamide or abiraterone/prednisone for patients with metastatic castration-resistant prostate cancer (mCRPC) (Phase Ib/II), with prednisolone for patients with DLBCL or FL (Phase II), and with pembrolizumab for patients with advanced/metastatic urothelial carcinoma (Phase I/II).
The second EZH2 inhibitor to progress to clinical evaluation was GSK126 developed by GlaxoSmithKline (GSK) Unlike tazemetostat, GSK126 is administered intravenously GSK126 was initially investigated in a phase I clinical trial for relapsed/reformatory DLBCL, transformed FL, other non-Hodgkin’s lymphomas, solid tumours and multiple myeloma Unfortunately this clinical trial was halted due to insufficient evidence of clinical activity In 2019, researchers reported their hypothesis for the lack of efficacy They claimed the tumour microenvironment is crucial to the action of EZH2 inhibitors, as anticancer activity was evident when GSK126 was administered to immunodeficient (but not immunocompetent) hosts Researchers administered GSK126 to immunocompetent hosts (i) with depleted myeloid-derived suppressor cells (MDSC) or (ii) in combination with MDSC-depleting drugs gemcitabine and 5-fluorouracil With both drugs, a rescue of the anticancer effect was observed, strengthening their hypothesis that administration of GSK126 resulted in increased levels of MDSC leading to diminished CD8+ T-cell response The authors claim that GSK126 suppresses the anti-tumour immune response which offsets the anti-proliferative effects.
At the time of writing, five other EZH2 inhibitors were under evaluation in clinical trials Constellation Pharmaceuticals had two EZH2 inhibitors in phase I/II trials Their first generation EZH2 inhibitor CPI-1205 is a novel substituted indole derivative CPI-1205 is an orally bioavailable drug which had completed a phase I study of patients with B-cell lymphomas Results showed anti-cancer activity and no significant toxicity issues CPI-1205 also produced positive phase Ib results for patients with mCRPC, demonstrating good tolerability, minimal safety concerns and promising clinical activity However, the data from the phase II ProSTAR study which evaluated CPI-1205 in combination with enzalutamide or abiraterone/prednisone for mCRPC did not show the required activity to progress the inhibitor further in this setting A second trial evaluating CPI-1205 targets patients with advanced solid tumours in combination with the anti-CTLA-4 antibody ipilimumab Researchers claimed that EZH2 inhibition modifies the function of regulatory T-cells, promoting an effector-like T-cell response which leads to increased anti-tumour immunity Additionally, Constellation Pharmaceuticals claimed that anti-CTLA-4 therapies lead to upregulation of EZH2, potentially counteracting their effectiveness Thus, Constellation had shown that the combination of EZH2 inhibition with the anti-CTLA4 agent ipilimumab enhances efficacy in tumour-bearing mice, providing a rationale for combining both therapies.
More recently, Constellation Pharmaceuticals described a second generation of EZH2 inhibitors According to these reports, replacing the methoxy substituent on the pyridone with a thiomethyl group resulted in significantly longer residence times However, this modification was also associated with increased CYP3A4 expression Through medicinal chemistry optimisation, Constellation developed compound 21 which combines good potency, a longer residence time and pharmacokinetics suitable for clinical development The compound displayed >91% TGI at a dose of 25 mg/kg BID-PO for 25 days in a Karpas-422 lymphoma mouse model Constellation’s compound 21 and related chemical equity are included in a patent from 2019, while a second patent from the same year describes compounds featuring the thiomethyl pyridone motif These compounds contain a 1,3-benzodioxole core similar to that of the dual EZH1/EZH2 inhibitors reported by Daiichi Sankyo (such as valemetostat) In contrast to the Daiichi Sankyo compounds which engage both EZH1 and EZH2, Constellation had not presented evidence for EZH1 inhibition Constellation initiated a phase I trial for their second generation inhibitor CPI-0209 for patients with advanced solid tumours CPI-0209 is a reversible inhibitor reported to have sub-pM PRC2 binding affinity and a residence time of longer than 3 months Additionally, Constellation Pharmaceuticals claimed that CPI-0209 has improved in vitro and in vivo potency in hematologic malignancies and solid tumour models compared to CPI-1205, tazemetostat and GSK126 Following the outcome of the ProSTAR study for CPI-1205, Constellation Pharmaceuticals announced that they were prioritising the development of CPI-0209.
Daiichi Sankyo developed the EZH2 inhibitor valemetostat (DS-3201), which was under evaluation in five clinical trials The compound is an orally available 1,3-benzodioxole derivative featuring a chiral spiroketal core In contrast to previous agents, valemetostat is a dual EZH1 and EZH2 inhibitor Although EZH1 activity was initially hypothesised to pose a potential safety risk, researchers tested dual EZH1 and EZH2 inhibitors and observed increased potency with no significant toxicity when tested in rats for 14 days Valemetostat recently received ‘Sakigake’ designation for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma following successful preliminary results of their phase I clinical trial for patients with non-Hodgkin’s lymphoma This designation, which is intended to expedite the approval of innovative medicines initially developed in Japan, signifies that valemetostat will have prioritised consultation as well as having the review time decreased from 12 to 6 months.
SHR2554 is an orally bioavailable EZH2 inhibitor patented by Jiangsu Hengrui Medicine currently in two clinical trials Little information has been revealed to date, and the chemical structure remains undisclosed However, a recent patent application describes novel benzofuran derivatives featuring a frequently-used pyridone moiety; a representative structure is example 3 with an IC50 of 5.7 nM.
Pfizer is the final company with an EZH2 inhibitor in the clinic PF-06821497 is currently being evaluated in a phase I trial Pfizer’s compound differs from its competitors in containing a lactam moiety, which the authors claim increases potency The inventors suggest that this could be due to decreased strain and/or improved contacts with the protein The lactam was initially accompanied by poor metabolic stability and thermodynamic solubility which necessitated further optimisation to develop PF-06821497, for which Pfizer claims high potency and good drug-like properties.
EZH2 Inhibitors in the Patent Literature 2016-2020
In addition to the six EZH2 inhibitors currently in clinical trials, new patent applications for EZH2 inhibitors have continued to emerge All of the compounds featured in this section bind to the EZH2 SAM binding domain and contain the methylpyridone structural motif Research groups have explored a wide variety of chemical modifications with different profiles; some compounds are EZH2-selective inhibitors whilst others are dual EZH1 and EZH2 inhibitors In addition, two applications describe irreversible EZH2 inhibitors.
GSK continued to publish patents describing various series of EZH2 inhibitors Following on from their thiophene-3-carboxamide derivatives described in 2016, GSK reported thieno[3,2-c]azepin-4-one derivatives in 2017 These compounds bear close similarity to those presented in the 2016 application, but with the central amide cyclised to form an azepinone ring The 2017 application describes 11 examples, all of which display IC50 values below 10 nM in a biochemical scintillation proximity assay (SPA) An additional patent application featuring different substitution patterns on the piperidine ring has since been disclosed
The 2016 and 2017 GSK applications bear some structural similarity to those published by Eli Lilly In 2016, Eli Lilly initially described two thiophene carboxamide derivatives This application includes example 1, which displays IC50 values of 0.93 nM against EZH2 WT and 1.43 nM against the Y641N EZH2-mutant in a biochemical assay Echoing the work by GSK, in 2017 researchers at Eli Lilly cyclised the amide to form a subset of 7-5 heterocycles Example 38 is claimed to have an IC50 of 0.92 nM against EZH2 WT and 2.65 nM against the Y641N EZH2-mutant Example 38 was further evaluated in vivo (A2780 xenograft models) in combination with ovarian cancer standard of care In these studies, the compound showed tumour growth inhibition of ΔT/C = 10.5% alone and 15.6% in combination with cisplatin and paclitaxel, whereas cisplatin and paclitaxel alone showed weak tumour inhibition with a ΔT/C value of 64.9%.
The replacement of the central amide motif with a lactam ring is featured in multiple applications Prior to the work disclosed by GSK and Eli Lilly, Pfizer developed a clinical candidate containing a 6-membered lactam ring In 2018, Mirati Therapeutics also disclosed a series of 3,4-dihydroisoquinolin-1-one derivatives featuring a lactam ring, such as example 17 for which the authors report an IC50 value of 1 nM in a methyltransferase assay.
Another structural element found in multiple patent applications is the benzamide motif The Dana-Farber Cancer Institute disclosed EZH2 inhibitors that include the benzamide moiety as a structural feature in both reversible and irreversible derivatives with dual EZH1 and EZH2 activity One such irreversible example is EZ-53, which is claimed to be a time-dependent and highly potent irreversible inhibitor of EZH1 and EZH2 The concept of covalent EZH2 inhibition was first described in 2016 Using X-ray crystallography, researchers identified Cys663, a cysteine residue close to the binding site, which the authors successfully targeted using an acrylamide covalent warhead.
Ancureall Pharmaceutical Co. Ltd also described a selection of irreversible benzamide containing EZH2 inhibitors These ligands are selective for EZH2 mutants A677G and Y641N over EZH2 WT or EZH1 Example 34 from this application displays a GI50 value below 10 nM against EZH2 A677G (Pfeiffer cell line) and below 100 nM against EZH2 Y641N (Karpas-422 cell line) In addition, Example 34 was also tested in vivo in mice for 27 days at a dose of 200 mg/kg in which the compound showed 67.1% tumour growth inhibition.
Another patent application describing benzamides was published by Tarapeutics Science Inc. and Hefei Institutes of Physical Science, Chinese Academy of Sciences featuring a range of reversible compounds exemplified by example 65, which has an IC50 of 24 nM measured in a Pfeiffer cell assay Example 68 disclosed in a recent application from Sinovent also contains the benzamide motif and contrasts with previous disclosures due to incorporation of a heteroatom at the 5 position of the benzamide Example 68 has an IC50 of 0.6 nM against wild-type EZH2 and has been tested in vivo in human B lymphocyte Pfeiffer subcutaneous xenograft tumour mice models showing 94.3% tumour growth inhibition at a dose of 100 mg/kg.
HaiHe Pharmaceuticals Co. Ltd and Shanghai Institute of Materia Medica of the Chinese Academy of Sciences also described a series of dual EZH1 and EZH2 inhibitors The authors disclose a series of indolizine carboxamides illustrated by example 2 Example 2 is claimed to have an IC50 of 1.39 nM against EZH2 Y641F mutant, with IC50 values against EZH2 WT or EZH1 not included in the patent application Prior to this, the same authors published another application describing fused pyridine EZH2 inhibitors In contrast to their newer publication, the authors did not claim EZH1 inhibition The application describes a number of compounds which have IC50 values below 100 nM.
Prior to the Dana-Farber Cancer Institute, HaiHe Pharmaceuticals Co. Ltd and Daiichi Sankyo, the University of North Carolina (UNC) was the first organisation to report an orally bioavailable dual EZH1/EZH2 inhibitor Researchers at the UNC developed UNC1999 which, in contrast to the compounds discussed previously, has been reported in the scientific literature rather than featured in a patent application The compound features a 22-fold selectivity margin for EZH2 over EZH1.
Following on from their 2016 application, Bayer described novel quinoline derivatives that inhibit EZH2 Example 3 is claimed to have an IC50 of 13 nM against EZH2 WT and 1.29 μM against EZH2 Y641N mutant in SPA experiments Yangtze River Pharmaceutical Group and Shanghai Haiyan Pharmaceutical Technology Co. Ltd, who have applied for numerous patents in this research area, disclosed isoquinoline derivatives in 2018 Z-138 is an example of such an isoquinoline and displayed an IC50 of 0.096 μM against EZH2 in a Pfeiffer cell proliferation assay Three other patent applications for EZH2 inhibitors have been filed by the same group in the past two years New heterocyclic derivatives with increased potency compared to the isoquinoline derivatives are disclosed in one application These compounds consist of a 3-methylindole core substituted with a carboxamide at the 4 position, similar to that of GSK-126, but with multiple areas of distinction including the sulphonyl substituent on the NH of the indole G-20 is claimed to have an IC50 value of 0.012 μM in the Pfeiffer cell assay A third application from 2018 describes indazole derivatives P-19 is claimed to have an IC50 of 0.076 μM in the Pfeiffer cell assay and is also a main focus of a 2019 application which describes polymeric forms of EZH2 inhibitors.
Resistance to EZH2 Inhibitors
Following the approval of tazemetostat for the treatment of ES and with many other EZH2 inhibitors making their way through the clinic, drug resistance is emerging as a potential concern Researchers engineered GSK126 resistant DLBCL cells in order to study mutations involved in the resistance to EZH2 inhibitors The researchers identified the mutations and tested the effects of these on the binding of both GSK126 and tazemetostat The study showed that the EZH2C663Y and EZH2Y762F mutants were resistant to both inhibitors In these cell lines, GSK126 and tazemetostat failed to inhibit DLBCL cell growth and a decreased level of apoptosis was observed compared to non-resistant cell lines (EZH2WT and EZH2E720G) They also studied the effects of the EZH2 inhibitor UNC1999 on these cells, as this compound does not directly interact with amino acid 663 As expected, UNC1999 was not affected by resistance in the EZH2C663Y mutant setting and retained activity against these cells However, UNC1999 does interact with amino acid 762, and therefore resistance was observed for this mutation This study demonstrates that mutations in the binding site can decrease the effectiveness of EZH2 inhibitors as a cancer therapy, but that some inhibitors may retain efficacy depending on the setting This provides hope for a potential switch in therapies when resistance occurs, and provides an impetus for increased knowledge of resistance-driving mutations to inform the design of future inhibitors Researchers suggested that EZH2 inhibitors could be developed based on residues in the active site rather than interaction with the extended pocket, which might overcome resistance Additionally, the discovery and development of EED inhibitors and other modalities could provide an alternative approach to inhibiting the PRC2 complex when resistance occurs.
EED Inhibitors
EED Inhibitors in the Patent Literature 2016-2020
In recent years, many organisations have reported compounds binding to EED which allosterically inhibit PRC2 These compounds generally bind to the H3K27me3 peptide binding site of EED, which leads to a conformational change in the EED H3K27me3 binding site and subsequent disruption of the EED-EZH2 protein-protein interaction Despite binding to a site other than the catalytic SET domain of EZH2, EED ligands have been shown to modulate the histone methyltransferase activity of EZH2 and therefore provide a promising new modality to target PRC2.
Novartis has played a seminal role in the discovery of EED inhibitors and were currently the first and only company to enter the clinic with an EED-targeted therapy Thus, MAK683 is an EED binder currently in a Phase I/II study in patients with advanced malignancies in DLBCL, nasopharyngeal carcinoma (NPC) or other advanced solid tumours where no further effective standard treatment is available In June 2016, a first EED-related patent application from Novartis was published describing a series of triazolopyrimidines derivatives for use in treatment of disease mediated by EED and/or PRC2, including the methyl pyridine compound example 2a Example 2a is reported to inhibit EED with an IC50 of 0.06 μM in an EED AlphaScreen binding assay and 0.003 μM in a Karpas-422 antiproliferative cell assay A patent application from Novartis was later published claiming crystalline forms of example 2a, with a specific focus on the hydrochloride salt.
In early 2017 Novartis disclosed a number of tool compounds identified from a biochemical high throughput screening (HTS) campaign which inhibit PRC2 activity through binding to EED The HTS output included a variety of different chemical scaffolds (EED162, EED396, EED666, EED709, and EED210), all of which yielded crystal structures bound to the trimethyl lysine pocket of EED and revealed an induced-fit pocket formed through conformational changes of the aromatic cage residues Phe97, Trp364 and Tyr365 which otherwise bind to the trimethylated lysine residue of histone H3 (H3K27me3) All five hit molecules were able to fully antagonise the binding of the H3K27me3 peptide to EED in the AlphaScreen competition binding assay with activities lower than 12 μM, with the two most potent compounds being EED210 (0.89 μM) and EED162 (0.32 μM).
A subsequent campaign of structure-guided design using the crystal structure of EED210 led to the discovery of compound 19 via deconstruction of the tricyclic core to afford a fragment-sized molecule with increased ligand and lipophilic efficiency (LE and LipE), followed by optimisation toward the featured bicyclic core Compound 19 incorporates a fluoro-substituted methoxyphenyl moiety which binds within the induced-fit pocket of EED and exhibits cellular activity of 2.9 μM in a G-401 ELISA assay Novartis also pursued a structure-guided approach using EED162 as a starting point leading to the discovery of tool compound EED226 as a first-in-class, potent and orally bioavailable inhibitor of EED In this campaign, EED162 was initially trimmed back to the triazolopyridine core and then regrown to increase potency, followed by a scaffold hop to the triazolopyrimidine core The triazolopyrimidine 6-aza component replaces the cyano substituent at position 6 in their original hit Finally the addition of a property-modulating sulphone group led to EED226 which had a cellular potency of 0.22 μM in the G401 ELISA assay and 0.08 μM in the Karpas-422 antiproliferative assay Furthermore, EED226 has been shown to induce complete tumour regression in a subcutaneous Karpas-422 xenograft model in vivo when dosed at 40 mg/kg for 32 days and showed evidence of sustained inhibition in cancer cell lines with acquired resistance to SAM-competitive EZH2 inhibitors.
In addition to EED226, Novartis pursued additional scaffolds including the triazolopyridine and imidazopyrimidine cores featured in two further patent applications A representative example from the triazolopyridine application is example 2b with an IC50 of 0.0109 μM in an AlphaScreen binding assay The imidazopyrimidine patent is exemplified by example 25 Example 25 features the same fluoro-substituted dihydrobenzofuran group from example 2a and has an IC50 of 0.007 μM in an AlphaScreen binding assay This application also explores a range of substituents at the 1-position of the imidazopyrimidine core, including but not limited to cyano, chloro, esters and small aromatic rings, several of which retain potency in an AlphaScreen binding assay.
In parallel to the work by Novartis, a collaboration between AbbVie and the Structural Genomics Consortium (SGC) independently reported the discovery of EED ligands exemplified by A-395 which is featured in a corresponding patent application for indane-containing EED inhibitors A-395 evolved from EED709 which was independently identified in a high throughput thermal shift screen Exploration of structure-activity relationships and structure-based design led to the cyclisation of the benzylamine moiety of EED709 to reveal the indane; these studies suggested the N,N-dimethyl moiety was critical for activity and demonstrated that a phenyl piperazine bearing a methylsulfonamide could provide additional potency as a replacement for the indole motif The published crystal structures for A-395 and EED226 show that both compounds bind to the same site and as with the Novartis HTS EED equity induce a reorganisation of the aromatic cage motif to reveal an induced-fit pocket A-395 exists as a mixture of two stereoisomers (epimeric at the indane stereocentre) and inhibits the catalytic activity of PRC2 in vitro with IC50 values of 18 nM and 90 nM in a radioactivity-based assay and a H3K27 methylation cell assay, respectively In addition, the compound exhibits antitumor efficacy in a DLBCL Pfeiffer xenograft model at 300 mg/kg subcutaneous dose two times per week for five weeks and retains potency in cell lines resistant to SET domain targeting EZH2 inhibitors.
In September of 2017 Fulcrum Therapeutics Inc. published a single compound application claiming compound 1 as a structurally-related EED inhibitor for the treatment of Fragile X syndrome (FXS) and other FMR1-inactivation-associated disorders The Fulcrum compound inhibits EED with an IC50 of 36 nM in an immunocytochemistry cell assay In patients with FXS, the FMR1 gene becomes transcriptionally silenced leading to a shortage in expression of Fragile X mental retardation protein (FMRP) which subsequently results in symptoms of FXS One potential treatment for FXS and other FMR1-inactivation-associated disorders is to reactivate the FMR1 gene Inhibition of EED subsequently inhibits the repressive function of PRC2 on the FMR1 gene, resulting in reactivation of the FMR1 gene.
In 2019 the Shanghai Institute of Materia Medica disclosed triazolopyrimidine and triazolopyridine derivatives for the treatment of PRC2 mediated disease through EED inhibition The first of two patent applications exemplifies a series of triazolopyrimidines with a variety of carbon-linked rings at the 8 position of the core as exemplified in E-Y13 which inhibits EED with an IC50 of 0.019 μM in a PRC2 enzyme assay The second application features the same triazolopyrimidine core, exploring nitrogen-linked saturated rings in the same position as demonstrated in SL-ZYE-36 which inhibits EED with an IC50 of 0.003 μM in a PRC2 enzyme assay.
Mirati also published an EED inhibitor application in 2019 that details a series of imidazopyrimidines exemplified by example 86 which demonstrates an IC50 of ≤ 100 nM in an ELISA cell-based assay and an IC50 ≤ 250 nM in a Karpas-422 anti-proliferative assay The Mirati application again demonstrates a wide variety of substituents at the 8 position of the core, comprising both carbon and nitrogen-linked aryl and heteroaryl groups.
In June 2019 Shanghai Blueray BioPharma Co. disclosed a series of EED ligands with a pyrimidone core as exemplified in BR-001 An x-ray crystal structure confirms BR-001 binds in the methyl lysine pocket of EED, and further experiments suggest this compound is also able to modulate the tumour microenvironment BR-001 has displayed phenotypic activity in vitro with robust anti-tumour activity in both Karpas-422 and Pfeiffer xenograft mouse models In addition, 30 mg/kg oral dosing of BR-001 led to 59.3% tumour growth suppression in a syngeneic CT26 colon tumour-bearing mouse model A second application from Shanghai Blueray BioPharma Co. published later in 2019 covers a number of 6,6 bicyclic cores, focussing primarily on a pyridopyrimidine core as exemplified by compound 17 A third application from the same authors published in early 2020 covers a number of 5,6 fused bicyclic cores such as compound 3; additional examples showcase an additional triazolopyrazine core and a thiadiazolopyridine core.
In 2020 the University of Jinan published two applications describing two distinct chemical compounds, compound 35 and compound 49, which were identified by virtual screening and subsequent biological evaluation Both compound 35 and 49 showed inhibition of the EED-EZH2 interaction with IC50 values of 64 and 55 μM respectively, complemented by anti-proliferative activity against Toledo DLBCL cancer cells.
EED Inhibitors Reported in the Non-Patent Literature 2016-2020
In addition to the patent literature, there have been several reports of EED ligands in peer-reviewed scientific journals The SGC in collaboration with the UNC Centre for Integrative Chemical Biology and Drug Discovery and the Princess Margaret Cancer Centre and Department of Medical Biophysics has been investigating peptidomimetic ligands of EED for a number of years In early 2017 this group revealed a number of peptidomimetic EED inhibitors including UNC4859 and UNC5114 discovered through a combined approach of combinatorial chemistry and structure-based design Each of these ligands afforded 80% inhibition of PRC2 catalytic activity in a methyltransferase SPA assay Furthermore, biotinylated UNC5114 enabled chemoprecipitation experiments which demonstrated that UNC5114 engages endogenous EED without disrupting the PRC2 complex In contrast to the majority of EED inhibitors which induce a change to the binding pocket of EED, these peptidomimetic ligands adopt a binding mode analogous to the endogenous EED ligands.
In 2019 AstraZeneca published the discovery of a series of EED binding compounds featuring a thiazole core This series was initiated through virtual screening of a commercial library against the EED protein structure, followed by structure-based drug design and medicinal chemistry optimisation The lead compound AZ1 demonstrates an IC50 of 18 nM against EED in a biochemical binding assay and shows antiproliferative activity in Karpas-422 cells with a GI50 of 0.63 μM Furthermore, AZ1 exhibits oral bioavailability and demonstrates in vivo efficacy in a Karpas-422 xenograft mouse model.
In 2020, scientists at Cardiff University published the discovery of EED degrading PROTACs (PROteolysis TArgeting Chimeras) These bifunctional molecules comprise an EED binding moiety linked to an E3 ligase recruiting ligand The lead PROTAC UNC6852 demonstrates potent and selective degradation of EED with a DC50 of 0.05 μM in HeLa cells Importantly, UNC6852 induces degradation of the entire PRC2 complex and demonstrates antiproliferative effects in multiple cancer cell lines This represents a novel approach to target PRC2 through protein degradation rather than direct inhibition.
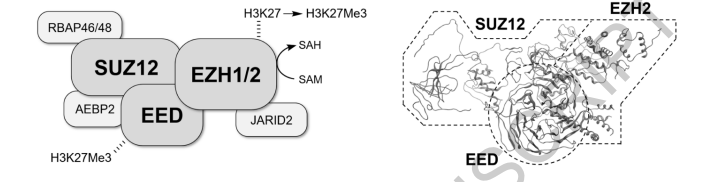
Fig. 2. EZH2 inhibitors under clinical evaluation or included in clinically-related patent literature.
PRC2 Activators
While the majority of research has focused on PRC2 inhibition, there have been limited reports on PRC2 activating compounds These compounds aim to target cancer cells with loss-of-function mutations where restored PRC2 activity may provide therapeutic benefit In 2018, researchers reported the identification of PRC2 activating compounds through a high-throughput screen The lead compound EML425 demonstrates the ability to increase H3K27me3 levels in cells and exhibits selective toxicity in PRC2-deficient cancer cell lines While still in early development, these activators represent an interesting complementary approach to PRC2 inhibition for specific cancer contexts.
PROTAC Degraders
Beyond EED degraders, researchers have also explored PROTAC approaches targeting other PRC2 components EZH2-targeting PROTACs have been developed that combine EZH2 binding elements with E3 ligase recruiting moieties These bifunctional molecules aim to induce targeted degradation of EZH2 rather than simply inhibiting its catalytic activity Early examples such as MS177 have demonstrated selective EZH2 degradation in cancer cell lines with corresponding decreases in H3K27me3 levels and antiproliferative effects This degradation approach may offer advantages over direct inhibition including the potential to overcome resistance mutations and target non-catalytic functions of EZH2.
Clinical Landscape and Future Directions
The approval of tazemetostat has validated EZH2 as a druggable target for cancer treatment, opening the door for additional PRC2-targeting therapies Several other EZH2 inhibitors are advancing through clinical trials including valemetostat with its dual EZH1/EZH2 activity and CPI-0209 with its extended residence time MAK683 represents the first EED-targeting therapy to enter clinical evaluation and will provide important insights into this alternative approach The diversity of mechanisms being explored including direct inhibition, allosteric modulation, protein degradation, and even activation provides multiple avenues to target PRC2 function.
Resistance mechanisms are already emerging for EZH2 inhibitors including specific mutations that reduce drug binding affinity The development of alternative approaches such as EED inhibitors and PROTAC degraders may help address these resistance challenges Furthermore, combination approaches with other targeted therapies or immunotherapies may enhance efficacy and overcome resistance.
Expert Opinion
The development of PRC2-targeting therapies has progressed rapidly from early tool compounds to FDA-approved medicines within the past decade The approval of tazemetostat marks the clinical validation of EZH2 as a cancer target and provides proof-of-concept for epigenetic cancer therapy However, several challenges remain that must be addressed as the field advances.
The limited efficacy observed with some EZH2 inhibitors such as GSK126 highlights the importance of understanding patient selection and combination approaches The tumor microenvironment appears to play a crucial role in determining response to PRC2 inhibition, and strategies to modulate immune suppressive factors may enhance efficacy Additionally, the identification of predictive biomarkers beyond EZH2 mutations will be critical for selecting patients most likely to benefit from PRC2-targeted therapy.
The diversity of approaches being explored provides multiple opportunities to address different aspects of PRC2 biology EED inhibitors may offer advantages in cancers driven by non-catalytic EZH2 functions, while PROTAC degraders could potentially overcome resistance mutations that affect direct inhibitors Dual EZH1/EZH2 inhibitors such as valemetostat may provide enhanced efficacy through broader target coverage.
Looking forward, the combination of PRC2 inhibitors with other targeted therapies, immunotherapies, or epigenetic modifiers represents a particularly promising avenue The interplay between PRC2 function and immune surveillance suggests that combining PRC2 inhibitors with checkpoint inhibitors or other immunomodulatory agents may enhance therapeutic outcomes Additionally, the development of more selective inhibitors and improved drug delivery approaches may help optimize the therapeutic window for PRC2-targeted therapies.
The patent landscape reveals continued innovation in PRC2 inhibitor design with numerous organizations pursuing novel chemical scaffolds and mechanisms of action This competitive environment should drive continued advancement in the field and provide multiple therapeutic options for patients As our understanding of PRC2 biology continues to evolve, new opportunities for therapeutic intervention will likely emerge.
Conclusions
The targeting of PRC2 for cancer treatment has evolved from a promising research area to clinical reality with the approval of tazemetostat The diversity of approaches being pursued including direct EZH2 inhibition, allosteric EED modulation, protein degradation, and complex activation provides multiple avenues to modulate PRC2 function While challenges remain including resistance development and patient selection, the continued innovation in this space promises to deliver additional therapeutic options for cancer patients The wealth of compounds in development represents significant progress toward realizing the full therapeutic potential of PRC2-targeted cancer therapy.